Guzy naczyniowe i wrodzone malformacje naczyniowe
Wprowadzenie i klasyfikacja
Rozwojowe zmiany naczyniopochodne są najpowszechniej występującymi zaburzeniami rozwojowymi. Stanowioną heterogenną grupę zmian, zarówno pod względem morfologicznym, jak i szerokim spektrum manifestacji klinicznej. Mogą objawiać się jako dyskretne, często przemijające, defekty kosmetyczne; lecz także powodować, ze względu na rozmiary, lokalizację oraz przebieg kliniczny, poważne skutki czynnościowe, aż do bezpośredniego zagrożenia życia. Zmiany naczyniopochodne mogą również stanowić składową mnogich zespołów chorobowych.
Występowanie wrodzonych zmian naczyniopochodnych i spowodowane nimi widoczne zniekształcenia były powodem poszukiwania przyczyn ich powstania. Od wieków wierzono, że są one skutkiem matczynych doznań z okresu ciąży, gdzie bodziec mogły stanowić zarówno wrażenia wzrokowe, przeżyte emocje, jak i myśli i marzenia. Tradycyjnie postrzegano je jako formę „naznaczenia”, utożsamiając ich powstanie z domniemanym przewinieniem matki w okresie ciąży. Atmosfera mistycyzmu i niedopowiedzenia miała wpływ na używaną nomenklaturę, często przypominającą język mitów i legend. Częściowo z tego powodu do dnia dzisiejszego; zarówno w języku potocznym, jak i słownictwie medycznym; funkcjonuje wiele określeń mających charakter czysto opisowy, niezwiązanych z faktycznym pochodzeniem czy charakterem biologicznym zmiany (np. „naczyniak o typie truskawki”, „znamię winnej plamy” czy „plama łososiowa”). [4]
W XIX wieku R. Virchow i G. Wegner podjęli próbę uporządkowania nomenklatury, wprowadzając klasyfikację anatomopatologiczną w oparciu o obrazy mikroskopowe. Podzielili oni ogromną i zróżnicowaną grupę zmian naczyniopochodnych na płaskie, proste, jamiste, torbielowate itd. Stworzony system znalazł praktyczne zastosowanie przez wiele lat. W dalszym ciągu jednak nie pozwalał on na zróżnicowanie „naczyniaków” w zależności od ich charakterystycznych cech biologicznych, uwzględniając w klasyfikacji wyłącznie elementy opisowe. Pojęcie „naczyniak” pozostawało uniwersalnym określeniem dla wszystkich zmian naczyniopochodnych, bez względu na rodzaj tworzących je naczyń, czas i przyczynę powstania, a przede wszystkim przewidywane zachowanie biologiczne. Nazywano tak zarówno twory typowe dla okresu wczesnodziecięcego, cechujące się nieuniknioną samoistną regresją; jak również pozostałe „guzy” o pochodzeniu naczyniowym, które tej cechy nie posiadają. [4]
Klasyfikacja biologiczna.
Dopiero J.B. Mulliken i J. Glowacki w 1982 przedstawili „Klasyfikację biologiczną zmian naczyniowych skóry i tkanek miękkich”, opartą w pierwszym rzędzie na cechach biologicznej aktywności zmian naczyniopochodnych, a pomijającą opis wyglądu zewnętrznego. Wyróżnione zostały dwie grupy zmian:
- naczyniaki (najczęściej występujące łagodne guzy pochodzenia naczyniowego u dzieci, proliferujące w okresie niemowlęcym i podlegające spontanicznemu zanikowi przez okres dzieciństwa)
- malformacje naczyniowe (różnorodne zmiany zbudowane z dysplastycznych naczyń, zazwyczaj z dominującym jednym rodzajem lub kalibrem naczyń: włosowatych, żylnych, tętniczych i limfatycznych)
| Naczyniaki | Malformacje naczyniowe |
|---|---|
| Faza proliferacyjna Faza inwolucyjna | Włosowate CM Limfatyczne LM Żylne VM Tętnicze AM Mieszane CLM CVM LVM AVM |
Wprowadzenie powyższej kwalifikacji częściowo ułatwiło różnicowanie wrodzonych zmian naczyniowych w oparciu o przewidywalny przebieg naturalny, kinetykę komórkową, oraz ich budowę histologiczną. Stworzono wspólny język dla środowiska medycznego zajmującego się diagnostyką i leczeniem wrodzonych anomalii naczyniowych. [8]
W 1996 roku podczas warsztatów zorganizowanych przez International Society for the Study of Vascular Anomalies (ISSVA) zmodyfikowano podział zmian naczyniopochodnych rozszerzając grupę pierwszą o dwie kolejne jednostki chorobowe: tufted angioma (angioblastoma) (TA) i kaposiform hemangioendothelioma (KHE) [2]. Są to rzadko występujące, łagodne guzy pochodzenia naczyniowego, zazwyczaj pojawiające się w okresie niemowlęcym lub wczesnodziecięcym (wyjątkowo wrodzone), o podobnej do naczyniaków tendencji do proliferacji. Różnią je od naczyniaków charakterystyczne obrazy histologiczne oraz mniej przewidywalny naturalny przebieg; mogą one zarówno ulegać spontanicznej inwolucji, jak też stopniowo powiększać się wraz z upływem czasu. Charakterystycznym dla obu tych guzów jest częste współistnienie fenomenu Merritt-Kasabacha(KMP),czyli nasilonej koagulopatii ze znaczną trombocytopenią /mniej niż 10000 płytek/mm3/ związanej z zatrzymaniem płytek wewnątrz guza; powikłania kończącego się zgonem w około 20% przypadków pomimo intensywnego leczenia. KMP dawniej wiązany z rozległymi naczyniakami w fazie proliferacji jest obecnie uważany za typowy wyłącznie dla TA i KHE. Chociaż poziom fibrynogenu u tych dzieci może spadać a czas protrombinowy wydłuża się, występuje to później i jest pierwotnie związane ze zużyciem płytek.
Po warsztatach ISSVA w roku 1996 zmieniono nazwę grupy z „naczyniaki” na „guzy naczyniowe”. Od tamtej pory do grupy „guzów naczyniowych” zaliczane są następujące zmiany:
- Naczyniak wczesnodziecięcy (Hemangioma of infancy)
- Zrazikowy naczyniak włosowaty (pyogenic granuloma)
- Szybko zanikający naczyniak wrodzony (RICH)
- Niezanikający naczyniak wrodzony (NICH)
- Tufted angioma (angioblastoma Nakagawy)
- Kaposiform haemangioendothelioma
- Wrodzony endokrynny potworniak naczyniowy
- Wrzecionowatokomórkowy hemangioendothelioma
Nomenklatura w grupie malformacji naczyniowych pozostała niezmieniona. [5]
Cechy różnicujące naczyniaki (guzy naczyniowe) i malformacje.
W przypadku zdecydowanej większości anomalii naczyniowych do postawienia wstępnego rozpoznania, a tym samym zaplanowania leczenia i przedstawienia prognozy, wystarczy szczegółowy wywiad i badanie fizykalne. W niektórych przypadkach konieczna może być obserwacja zachowania zmiany na przestrzeni pewnego okresu czasu albo wykonanie dodatkowych badań obrazowych lub biopsji. [7] Warto jednakże pamiętać, że zmiany naczyniopochodne o całkowicie odrębnym pochodzeniu, biologii i przebiegu naturalnym mogą wyglądać łudząco podobnie. [4]
„Nie wszystkie zmiany skórne wyglądające jak truskawka to naczyniaki; nie wszystkie naczyniaki przypominają wyglądem truskawkę.” J.B. Mulliken, MD
Naczyniaki to guzy charakteryzujące się wysoką aktywnością metaboliczną; ze wzmożoną wymianą komórek śródbłonka, komórek tucznych, fibroblastów i makrofagów w okresie proliferacji objawiającą się zmianami wielkości, kształtu, zabarwienia i spoistości; z nieuchronnie następującą fazą inwolucji, podczas której na skutek zmniejszenia aktywności metabolicznej guza dochodzi do jego stopniowego zaniku.
Malformacje naczyniowe są guzopodobnymi zmianami nienowotworowymi, powstałymi na skutek zaburzenia procesu morfogenezy tkanki naczyniowej. Charakteryzują się normalnym cyklem wymiany komórkowej poprzez cały wszystkie fazy rozwoju i nie podlegają samoistnej inwolucji. Rodzaj utkania naczyniowego warunkuje podział malformacji naczyniowych na podkategorie (włosowate /kapilarne/, żylne, tętnicze i limfatyczne lub mieszane). Drugim parametrem określającym charakter zmiany jest prędkość przepływu krwi w jej obrębie. Wyróżnione zostały zmiany niskoprzepływowe (zawierające elementy naczyń żylnych, włosowatych i limfatycznych) i wysokoprzepływowe (z elementami tętniczymi). Pomimo, że są to zmiany wrodzone mogą pozostać „niemymi” klinicznie do okresu pokwitania i dorosłości. Najczęściej występują pojedynczo. W sporadycznych przypadkach udowodniono występowanie rodzinne (dwie rodziny z wrodzonymi malformacjami żylnymi z określoną mutacją w obrębie genu VMCM-1 zlokalizowanego na chromosomie 9p). [2]
| Naczyniak | Malformacja naczyniowa |
|---|---|
| Cechy kliniczne | |
| Pojawia się w okresie noworodkowym lub wczesnoniemowlęcym | Zawsze obecna w chwili porodu |
| Rośnie szybciej od dziecka przez 1 rok życia | Rośnie proporcjonalnie wraz z dzieckiem |
| Inwolucja po 1 roku życia | Nigdy nie zanika |
| Częstość występowania ♂:♀ 1:6 | Częstość występowania ♂:♀ 1:1 |
| Cechy biologiczne | |
| Proliferacja: obfite, proliferujące komórki śródbłonka, liczne komórki tuczne | Płaskie, normalne komórki śródbłonka, liczba komórek tucznych w normie |
| Inwolucja: Apoptoza i postępujące spłaszczanie komórek śródbłonka. Szersze, ale mniej liczne naczynia, otoczone okołonaczyniową tkanką włóknisto-tłuszczową | Normalna wymiana komórek śródbłonka; dysplastyczne kanały naczyniowe: żyłki, żyły, włośniczki lub naczynia limfatyczne |
Naczyniaki
Są najczęstszymi guzami okresu dziecięcego. Typowo są niewidoczne w chwili urodzenia, pojawiają się i zostają zauważone w ciągu pierwszych tygodni życia – dotyczy to około 70% wszystkich naczyniaków. Pierwszymi objawami mogą być: dyskretna płaska, różowa plama, plama wyblakła lub ograniczona teleangiektazja otoczona bladym halo. Kolejnych 20% dzieci rodzi się z widocznym już objawem prodromalnym, a naczyniak rozwija się w bardzo krótkim czasie po urodzeniu. W około 10% przypadków dojrzały naczyniak obserwowany jest już w chwili urodzenia. Są to tzw. naczyniaki wrodzone, często bez tendencji do proliferacji i z bardzo szybko przebiegającą fazą inwolucji (rapidly involuting congenital haemangioma - RICH), lub bez tendencji do zaniku (non-involuting congenital haemangioma – NICH). Niektórzy autorzy uważają te guzy za zupełnie odrębne od naczyniaków, za czym przemawiają różnice obrazów histologicznych. Naczyniaki zlokalizowane w głębszych warstwach tkanek, a sięgające powierzchownie skóry, tkanki tłuszczowej podskórnej lub położone w mięśniach mogą pozostawać bezobjawowe przez wiele miesięcy. Niekiedy powierzchowne zmiany są tylko „szczytem góry lodowej”, – gdy dominująca masa naczyniaka penetruje do głębiej położonych tkanek.
Naczyniaki występują częściej u dziewczynek niż chłopców – w stosunku nawet 6:1, w zależności od opracowania; częściej również u dzieci rasy białej, chociaż problem dotyczy dzieci każdej rasy i koloru skóry.
Są to zmiany tak powszechne, że nawet 10% - 12% białych niemowląt w wieku 12 miesięcy ma przynajmniej jeden z nich. U dzieci z bardzo niską urodzeniową wagą ciała (<1000g) spotykane są w około 23% - 3 razy częściej niż u noworodków dorodnych i urodzonych o czasie (dotyczy to zresztą częstości występowania wszystkich anomalii naczyniowych).
Naczyniaki występują najczęściej (w około 60%) w obrębie głowy i szyi, w 25% w obrębie tułowia, i w 15% na kończynach. Według innych danych naczyniaki lokalizują się w 70 –80% na twarzy i szyi a w 20% w innych częściach ciała. Osiemdziesiąt procent naczyniaków występuje jako zmiana pojedyncza, 20% przypadków stanowią mnogie guzy zlokalizowane w różnych rejonach ciała. Mnogim guzom w obrębie skóry towarzyszą częściej naczyniaki umiejscowione w obrębie trzewi - (w kolejności zmniejszającej się częstotliwości występowania) w wątrobie, płucach, i przewodzie pokarmowym. Rzadką lokalizacją są węzły chłonne, śledziona, pęcherzyk żółciowym, trzustka, nadnercza, opony mózgowo – rdzeniowe, mózg i rdzeń przedłużony. Zazwyczaj są one związane ze zmianami skórnymi.
Szczególną cechą naczyniaków jest ich gwałtowny wzrost w okresie noworodkowym, znacznie szybszy niż wzrost dziecka, rozpoczynający się w ciągu kilku tygodni po urodzeniu. Faza proliferacyjna trwa około 6 – 12, a nawet 18 miesięcy. Kluczową rolę w tej fazie odgrywają czynniki wzrostu takie jak podstawowy czynnik wzrostu fibroblastów (bFGF), czy naczyniowy śródbłonkowy czynnik wzrostu (VEGF); a także proteazy (urokinaza, kolagenaza typu IV) oraz np. cząsteczki adhezyjne. Komórki śródbłonka w danej lokalizacji przypominają i zachowują się raczej jak w okresie embrionalnym niż noworodkowym, namnażając się znacznie szybciej niż inne komórki. Naczyniak staje się guzem bogatokomórkowym. Zmiana w powierzchownych warstwach skóry staję się wyniesiona, jej powierzchnia staje się guzowata i nierówna, o jaskrawo-karmazynowej lub jasno-szkarłatnej barwie. W ciągu pierwszego roku życia stopniowo kolor pogłębia się.
Naczyniaki położone w głębszych warstwach skóry i tkanki podskórnej powodują jedynie nieznaczne uniesienie skóry i jej błękitnawe zabarwienie lub powstanie bladych naczyń telangiektatyczych bez zmiany zabarwienia otaczającej skóry. Naczyniaki w fazie wzrostu są w badaniu palpacyjnym spoiste; niemożliwe jest opróżnienie ich z krwi poprzez zastosowanie ucisku.
Około 10-12 miesiąca życia wzrost naczyniaka staje się proporcjonalny do wzrostu dziecka, co wskazuje na rozpoczęcie fazy inwolucji. Jest to poprzedzone pojawieniem się znacznej ilości komórek tucznych i wzrostem poziomu produkowanych tkankowych inhibitorów metaloproteinaz, co zapoczątkowuje proces zaniku. Apoptoza komórek śródbłonka powoduje stopniowy zanik naczyniaka. Guz staje się stopniowo bardziej miękki i podatny, a jego powierzchnia blednie, przestaje być lśniąca, przybierając matowy odcień i fioletowe zabarwienie, a następnie ewoluuje przybierając odcień cętkowano – popielaty na całej powierzchni zmiany, zaczynając od środka i stopniowo postępując do obrzeża. Zarówno początek procesu inwolucji, jak i jej szybkość są nie do przewidzenia. Statystycznie 30% naczyniaków zanika do końca 3 roku życia, 50% - do końca 5-go, 70% - do 7 lat, a w pozostałych przypadkach proces inwolucji trwa nawet do 16 roku życia. [3,4,5,8,11]
Istotnym jest precyzyjne rozumienie pojęcia inwolucji, czyli zaniku. Tylko około 40% naczyniaków „zanika” praktycznie bez pozostawienia zauważalnego śladu lub z minimalną pozostałością w pełni akceptowalną pod względem kosmetycznym. W pracy Finna i wsp. wykazano, że naczyniaki można podzielić na te, które zakończyły swój proces inwolucji do końca 6 roku życia, i te, w których zanik trwał dłużej niż 6 lat. W pierwszej grupie „tylko” u 38% dzieci na koniec procesu zaniku pozostał ślad pod postacią: włóknisto-tłuszczowego nadmiaru skóry, blizny czy też widocznych teleangiektazji. W grupie drugiej już tylko u 20 % pacjentów pozostałość po procesie zaniku była w pełni do zaakceptowania. [3,11]
(tabela - Oczekiwany wynik końcowy naturalnego procesu inwolucji naczyniaka)
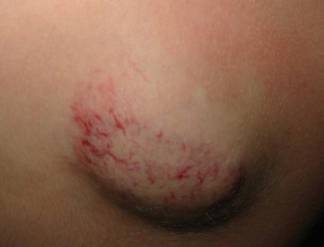
Naczyniak w początkowej fazie inwolucji
Okresem wymagającym szczególnej uwagi jest faza proliferacji naczyniaka. Jego gwałtowny wzrost, rozprzestrzenianie się w otaczających tkankach oraz kumulacja znacznej ilości krwi w jego obrębie wybitnie predestynują do powstania powikłań miejscowych lub ogólnych. Najważniejsze z nich to:
- Owrzodzenia występujące u mniej niż 5% dzieci z naczyniakami może doprowadzić do wtórnego zakażenia oraz niszczenia tkanek miękkich oraz chrząstek. Zmiany zlokalizowane w obrębie warg oraz okolicy narządów moczowo – płciowych są bardziej podatne na powstawanie owrzodzeń. Skutkiem wygojonego owrzodzenia jest blizna skórna.
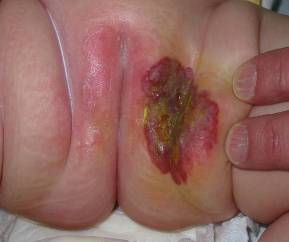
Naczyniak okolicy krocza powikłany owrzodzeniem
- Skutki ucisku lub współistnienia w okolicy naturalnych otworów ciała (szpary powiekowe, nozdrza, przewody słuchowe, usta, odbyt, cewka moczowa).
Naczyniak w obrębie głowy i szyi często zlokalizowany jest w obrębie powiek, grzbietu nosa, warg, okolicy przedusznej i wraz ze wzrostem może kolidować z naturalnymi otworami ciała, powodując ucisk lub zaburzenie ich funkcji.
Zmiany zlokalizowane w obrębie powiek, oraz oczodołu mogą powodować niedowidzenie, zez, wytrzeszcz, oraz wady refrakcyjne. Nawet stosunkowo niewielki twór w obrębie górnej powieki może doprowadzić do zniekształcenia rogówki i astygmatyzmu.
Podobnie zmiany umiejscowione w pobliżu ślinianki przyusznej, uciskając zewnętrzny kanał słuchowy doprowadzić mogą do upośledzenia słuchu.
Naczyniak podgłośniowy może powodować obturacje dróg oddechowych. Niebezpieczny dwufazowy stridor najczęściej występuje w wieku 2 – 3 miesięcy. Niekiedy objawy sugerują zapalne tło takie jak zapalenie krtani lub błonica. Dopiero rtg, KT albo bezpośrednia laryngoskopia szyi uwidacznia asymetryczny odruch połykania naprowadzający na rozpoznanie naczyniaka. Około 50% pacjentów z naczyniakiem podgłośniowym ma w obrębie twarzowo – szyjnym zmiany naczyniopochodne.
Naczyniaki warg utrudniają dzieciom odżywianie, powodują wady wymowy, a poprzez bezpośredni ucisk prowadzą do powstawania wad zgryzu.
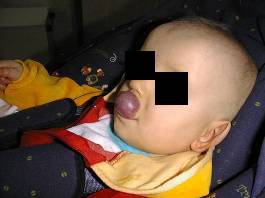
Naczyniak wargi górnej
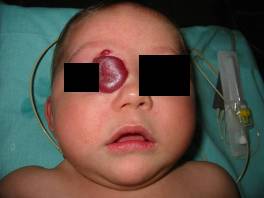
Naczyniak okolicy oczodołu
Naczyniaki położone w bezpośrednim sąsiedztwie krocza i narządów płciowych mogą zaburzać oddawanie moczu i stolca, pozostając szczególnie narażone na owrzodzenia i powikłania z tym związane.
- Zlokalizowany krwotok z powierzchni naczyniaka występuje rzadko z wyjątkiem towarzyszących zmian owrzodzeniowych. Uogólnione krwotoki zdarzają się w przebiegu zespołu Merritt- Kasabacha u noworodków z rozległymi guzami naczyniowymi (TA), (KHE). Śmiertelność w tym przypadku sięga 20 – 30% pomimo zastosowanego leczenia.
- Niewydolność serca z wysokim rzutem. Może wystąpić u dzieci z rozległymi mnogimi zmianami w obrębie skóry lub narządów wewnętrznych lub czasami w dużych guzach skórnych bez zajęcia innych narządów. Niewydolność serca spowodowana jest wypełnieniem krwią znacznych rozmiarów naczyń w obrębie guza naczyniowego oraz przeciekiem lewo-prawym. Dotyczy to najczęściej naczyniaków wątroby. Stwierdza się powiększenie wątroby oraz skurczowy szmer nad narządem. Dodatkowe powikłania: zakażenia, urazy (pęknięcia) oraz zespół Merritt-Kasabacha współodpowiadają za śmiertelność do 50% pacjentów z objawami zastoinowymi. [11]
Naczyniaki – leczenie
Szukając zaleceń dotyczących sposobów leczenia naczyniaków, najczęściej znajdziemy koncept, który można przedstawić następującym zdaniem:
Ze względu na swoją charakterystyczną aktywność biologiczną, większość naczyniaków (90%) nie wymaga żadnego leczenia.
Tak brzmi „aktualny standard” w leczeniu naczyniaków, oznaczając w znacznej mierze bierne oczekiwanie na nieuchronny, spontaniczny, ale też całkowicie nieprzewidywalny, proces inwolucji. Postępując zgodnie z tą zasadą akceptujemy z góry naturalnie zaplanowany przebieg fazy proliferacji, a następnie zaniku, decydując się na ewentualną ingerencję w skrajnych przypadkach lub po zakończeniu cyklu rozwoju zmiany. Wynika to z przekonania, że naturalny proces inwolucji zapewnia optymalne warunki, dając ostatecznie najlepszy wynik czynnościowy i kosmetyczny.
Czy aby na pewno tak powinno wyglądać współczesne podejście do leczenia tych niejednokrotnie szpecących i niosących konkretne powikłania zmian? Czy wraz z rozwojem medycyny nie posiadamy możliwości modulowania przebiegu poszczególnych faz rozwoju, powstrzymania proliferacji i wcześniejszego zainicjowania szybszej inwolucji?
Czy nie powinniśmy dążyć do uzyskania jak najlepszego wyniku funkcjonalnego i estetycznego, przy jednocześnie maksymalnym ograniczeniu negatywnych skutków w psychice dziecka, wynikających z bycia odmiennym?
Z całą pewnością powyższy „standard” nadal obowiązuje w odniesieniu do większości naczyniaków, dotycząc zmian małych, niepowikłanych, o miernej tendencji do wzrostu, wczesnym początku inwolucji; szczególnie tych zlokalizowanych w miejscach ukrytych takich jak tułów, skóra owłosiona głowy, czy kończyny z wyłączeniem rąk. Wymagają one tylko pielęgnacji w formie częstego natłuszczania oraz regularnych kontroli, najlepiej w poradni specjalistycznej.
Procesowi aktywnego leczenia podlegają natomiast wszystkie pozostałe zmiany, stanowiące potencjalne lub rzeczywiste zagrożenie poprzez zaburzenie funkcji lub powodowanie znacznego zniekształcenia. Zaliczamy do nich naczyniaki duże, szybko rosnące, powikłane, oraz o szczególnej lokalizacji. Zdecydowana większość naczyniaków z tej grupy umiejscowionych jest w obrębie głowy i szyi; najczęściej na pograniczu obszarów zainteresowania różnych specjalności klinicznych, stanowiąc problem interdyscyplinarny w procesie diagnostyki i leczenia.
Dlatego słusznym wydaje się tworzenie przy dużych ośrodkach klinicznych wyspecjalizowanych wieloosobowych „zespołów ds. wrodzonych anomalii naczyniowych”, w których skład wchodzić powinni chirurdzy dziecięcy, chirurdzy plastyczni, laryngolodzy, okuliści, radiolodzy, onkolodzy dziecięcy i inni. [4]
Bezwzględne wskazania do leczenia naczyniaków stanowią:
- Bezpośrednie zagrożenie życia (niewydolność serca, obturacja dróg oddechowych, znacznego stopnia koagulopatia)
- Zaburzenie podstawowych funkcji życiowych, takich jak: oddychanie, odżywianie i wydalanie
- Interferencja z naturalnymi otworami ciała (np. zaburzenia w kształtowaniu zmysłów takich jak wzrok i słuch)
- Zagrożenie pozostawieniem trwałych skutków w bezpośrednim sąsiedztwie (powieka górna – zniekształcenie rogówki - astygmatyzm, warga – wada zgryzu itp.)
- Zagrożenie zaburzeniem funkcji motorycznych (ręka – procesy uczenia, stopa – poruszanie)
- Powikłania (krwawienie, owrzodzenie z odczynem zapalnym)
- Szybki wzrost i szczególne lokalizacje stwarzające zagrożenie znacznym defektem kosmetycznym (np. czubek nosa)
- Blizny i tkanki resztkowe po procesie zaniku; istotne zarówno z przyczyn funkcjonalnych jak estetycznych
Pozostaje cała bardzo liczna grupa chorych z naczyniakami, które nie podlegają powyższym kryteriom; gdzie wskazania do leczenia można by określić jako „względne”. Należą tu niewielkie, niepowikłane zmiany, bez szczególnej tendencji do wzrostu, a przede wszystkim o powolnym lub opóźnionym zaniku, położone w miejscu eksponowanym (twarz, szyja, dekolt, ręce). Tradycyjna postawa wyczekująca wydaje się być uzasadniona przez okres około 18-24 miesięcy. Właśnie w tym wieku pojawiają się pierwsze symptomy kształtującej się świadomości własnego wyglądu. Dziecko dostrzega swoją „odmienność” i nierzadko w krótkim przestaje ją akceptować. Jeśli dodamy do tego niepokój i stres narastający wśród członków najbliższej rodziny pacjenta, to celowym wydaje się ponowne rozważenie wskazań do leczenia. Warto pamiętać, że wraz z rozwojem i coraz szerszą dostępnością nowoczesnych technologii, takich jak farmakoterapia, laserowa fotokoagulacja, czy też praktycznie bezkrwawe usuwanie chirurgiczne naczyniaków, „upieranie się” przy biernym wyczekiwaniu jest coraz mniej uzasadnione; szczególnie, że odsetek inwolucji naczyniaków bez pozostawienia widocznego śladu maleje wraz z wiekiem, a świadomość „bycia odmiennym” może spowodować trwałe skutki w psychice dziecka i jego rodziny.
Istnieje kilka zasadniczych i uznanych metod leczenia naczyniaków. Można je ze sobą łączyć lub stosować sekwencyjnie w celu uzyskania jak najlepszych efektów miejscowych i ogólnych:
1. Farmakoterapia
- Kortykosterydy stosowane ogólnoustrojowo
- Kortykosterydy wstrzykiwane miejscowo (podawane do naczyniaka)
- Interferon α-2a stosowany ogólnoustrojowo
- Winkrystyna stosowana ogólnoustrojowo
2. Fotokoagulacja laserowa
- Śródtkankowa Nd-YAG (1064 nm)
- Przezskórna Nd-YAG (1064 nm)
- Przezskórna FLPD (flashlamp pumped-dye laser 585 nm)
3. Wycięcie chirurgiczne
- Częściowe (klasyczne/ z wykorzystaniem lasera)
-Całkowite (klasyczne/ z wykorzystaniem lasera)
4. Inne
- Krioterapia
Farmakoterapia
Sterydy kory nadnerczy
Podstawowym lekiem wykorzystywanym w leczeniu naczyniaków od połowy lat 60-tych są sterydy kory nadnerczy. W międzyczasie stały się one:
„Lekiem pierwszego rzutu w leczeniu naczyniaków stanowiących zagrożenie dla życia lub wzroku.”
Sterydy działają na naczyniaki w fazie proliferacji. Najczęściej stosowane są one ogólnoustrojowo w dawce od 2-3 miligramów na dobę w jednej dawce porannej; niektórzy autorzy proponują nawet dawki do 5 mg na dobę. Dobra odpowiedź na leczenie pozwala na stosowanie pełnych dawek przez 4 do 6 tygodni a następnie stopniowe ich zmniejszanie w zależności od wrażliwości, lokalizacji i dojrzałości zmiany. Wystąpienie powiększania naczyniaka z „odbicia” wymaga ponownego stosowania wysokich dawek przez okres 2 tygodni przed stopniowym ich odstawieniem. Nie należy podawać sterydów w okresie inwolucji. Pomimo bardzo długiej listy potencjalnych działań niepożądanych, obejmującej między innymi: opóźnienie wzrostu, owrzodzenie żołądka, nadciśnienie tętnicze czy immunosupresja, sterydy są chętnie stosowane ze względu na prostotę terapii i stosunkowo wysoki odsetek pozytywnych odpowiedzi klinicznych (od ok. 30% - Bartoshesky et all.; 1978, Enjolras et all.; 1990; aż po 93% - Sadan i Wolach; 1996).
Dla zminimalizowania ewentualnych skutków ubocznych stosowane są leki działające osłonowo na przewód pokarmowy. Jednocześnie sterydy są podawane przez jak najkrótszy czas, zwłaszcza przy braku efektu leczniczego.
Miejscowe ostrzyknięcie naczyniaka sterydami wprowadzono pierwotnie w roku 1970 dla uniknięcia skutków ogólnych sterydoterapii doustnej. Od tej pory jest to uznany i zaakceptowany sposób leczenia, zwłaszcza naczyniaków ograniczonych np. o lokalizacji okołooczodołowej. Najczęściej stosowane są jednocześnie dwa sterydy: triamcinolon w dawce 3-5 mg/ kg c.c. i betametazon w dawce 0.5-1.0 mg/ kg c.c. Dołączenie długodziałającego triamcinolonu utrwala natychmiastowy efekt działania bethametasonu. Niestety uzyskany skutek jest nietrwały i może wymagać powtórzenia iniekcji po około 6 tygodniach. Lista skutków niepożądanych zawiera między innymi: niedrożność tętnicy środkowej siatkówki zarówno oka po stronie zmiany, jak i drugiego; martwicę powieki i tworzenie blizn zanikowych. Opisano również skutki ogólne sterydów po ich zastosowaniu lokalnym, co powoduje, że metoda ma tyleż zwolenników, co oponentów.
Interferon
Interferon wprowadzony do praktyki klinicznej początkowo jako lek antywirusowy, okazał się mieć działanie hamujące migrację komórek śródbłonka, jak i innych etapów angiogenezy. Między innymi hamuje on czynniki wzrostu biorące udział w stymulacji wzrostu naczyniaka (ECGF, FGF). Działa on niezależnie od fazy proliferacji, czyli przez cały cykl życiowy naczyniaka. Najczęściej stosowany jest interferon α2a w dziennej dawce 3000000j/m² w postaci podskórnych zastrzyków. Działania niepożądane są w większości związane z przejściowym dyskomfortem związanym z bólami mięśni, stawów, stanami podgorączkowymi. Opisano niestety przypadki nieodwracalnego lub częściowo odwracalnego niedowładu spastycznego kończyn dolnych. Z tych przyczyn interferon pozostaje lekiem stosowanym wyłącznie w przypadkach bezpośredniego zagrożenia życia, lub wzroku, po uprzednim niepowodzeniu w leczeniu sterydami. Poważną przeszkodą w stosowaniu interferonu pozostają wysokie koszty leczenia.
Fotokoagulacja laserowa.
Fotokoagulacja śródtkankowa / przezskórna laserem Nd-YAG.
Wskazania do stosowanie laseroterapii – laser neodymowo-yagowy (Nd-YAG) są szerokie i obejmują większość stanów klinicznych w przebiegu naczyniaka:
- Lokalizację naczyniaka w obrębie oczu, nosa, uszu i ust
- Nawracające stany zapalne z owrzodzeniem i krwawienia z powierzchni naczyniaka
- Obecność zmian o charakterze defektu kosmetycznego, powodujących problemy emocjonalne dziecka (poczucie inności o negatywnym zabarwieniu, niższe poczucie własnej wartości, trudności adaptacyjne w przedszkolu i szkole).
Fotokoagulację śródtkankową stosuje się w przypadkach zmian z obecnością znacznej komponenty podskórnej. Leczenie u dzieci przeprowadza się w warunkach znieczulenia ogólnego. Światłowód wprowadzany jest do wnętrza naczyniaka poprzez otwór wykonany jałową igła o odpowiedniej wielkości. Wewnątrz tkanki guza wykonuje się serie krótkich (10 s) ekspozycji światła lasera. Po usunięciu światłowodu nie jest konieczne założenie szwów, na powierzchnie stosuje się opatrunek z maści antybiotykowej (detreomycyna). Niezwykle istotne jest zwrócenie w czasie zabiegu uwagi na:
- Ochłodzenie tkanek wokół zmiany tak by zminimalizować efekt termiczny promieniowania – szczególnie w okolicy gałki ocznej
- Zwrócenie uwagi by końcówka światłowodu nie znajdowała się zbyt blisko powłok, co grozi – nawet w przypadku powierzchniowego ochładzania – oparzeniem i martwicą skóry.
W przypadku tej metody efekt kosmetyczny u większości dzieci obserwowany jest już po pierwszym zabiegu. Należy poinformować rodziców, iż w pierwszych dniach po operacji widoczny będzie obrzęk i pozorne powiększenie zmiany, które to objawy ustępują w ciągu 7 – 10 od rozpoczęcia terapii. [11]
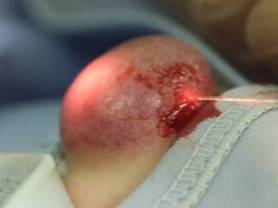
Światłowód wprowadzony do naczyniaka podczas fotokoagulacji śródtkankowej laserem Nd-YAG
Fotokoagulacja przezskórna laserem Nd-YAG.
Jest to forma laseroterapii stosowana w przypadku zmian położonych powierzchownie, z racji ograniczonej penetracji światła lasera w tkance (około 7-10 mm). Wymaga to posiadania specjalnego aplikatora oraz znieczulenia ogólnego w przypadku dzieci. Terapia odbywa się „poprzez czysty lód” używany w celu schładzania powierzchni i ochrony skóry. [9]
Fotokoagulacja przezskórna laserem FLPD.
Leczenie laserem o długości fali w zakresie światła żółtego, o maksymalnej absorbcji przez barwę czerwoną, pozwala na najbardziej wybiórczą fotokoagulację naczyń krwionośnych. Laser ten ma natomiast znacznie mniejszą zdolność penetracji w głąb tkanek, co ogranicza wskazania do jego zastosowanie wyłącznie do zmian położonych powierzchownie. Szczególnym wskazaniem pozostają teleangiektazje pozostające po zakończeniu procesu inwolucji. Ograniczeniem jest też znacznie mniejsza dostępność sprzętu.
Leczenie chirurgiczne
Leczenie chirurgiczne znajduje zastosowanie między innymi w następujących przypadkach:
- Zapewnienie dziecku prawidłowej drożności drogi oddechowych i pokarmowych w przypadku naczyniaka rozprzestrzeniającego się i uciskającego na ww. drogi
- Usunięcie zmiany z okolicy oczodołu, nozdrzy, odbytu
- Usunięcie pozostałości po naczyniaku, który zakończył fazę inwolucji
- Korekcja defektów czynnościowych np. zaburzeń ruchomości kończyny lub kosmetycznych
- Usunięcie małej dobrze ograniczonej zmiany
Najczęściej leczenie operacyjne połączone z wycięciem masy naczyniaka poprzedzone jest lasero- lub hormonoterapią.
Z racji lokalizacji większości naczyniaków w obrębie głowy i szyi, ze szczególną skłonnością do zajmowania warg, powiek, nosa wraz ze skrzydełkami, fałdów nosowo-policzkowych, policzka z pierwotnym związkiem ze ślinianką itd., stanowią one w wielu przypadkach problem plastyczno-rekonstrukcyjny z pogranicza chirurgii dziecięcej i plastycznej, laryngologii, okulistyki itd. Nierzadko ostateczny zabieg po okresie inwolucji, ale także etapy w trakcie leczenia wcześniej, wymagają od zespołu biegłości w zastosowaniu nowych technologii umożliwiających ograniczenie utraty krwi i zapewniających jak najlepszą widoczność w trakcie zabiegu (skalpel laserowy Nd-YAG), a także kompleksowego podejścia do rekonstrukcyjno-estetycznej części zabiegu.
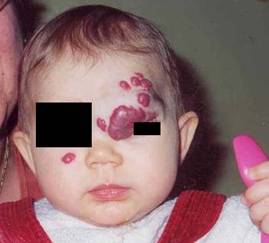
Naczyniak okolicy oczodołu upośledzający widzenie
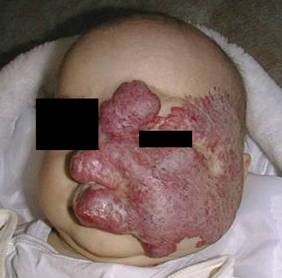
Masywny naczyniak lewej połowy twarzy
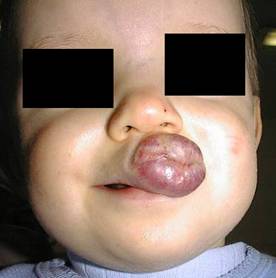
Naczyniak wargi górnej po 2 etapach leczenia laserem Nd-YAG
Malformacje naczyniowe
Malformacje naczyniowe charakteryzowane są przez dominujący typ kanałów naczyniowych tworzących zmianę. Często spotykane są formy mieszane. Niektóre mają charakterystyczne cechy i rozprzestrzenienie oraz towarzyszą innym zmianom o charakterze dysmorficznym, a także wchodzą w skład zespołów chorobowych. [5]
Klasyfikacja malformacji naczyniowych:
KAPILARNE (CM)
Znamiona winnej plamy
Zespół Sturge-Weber
Malformacje kapilarno-limfatyczne (CLM)
Ograniczone
„Angiokeratomy”
Teleangiektazje
Samoistna teleangiectazja
Zespół Rendu-Osler-Weber
Ataksja teleangiektazja – zespół Louis-Bar
Wrodzona teleangiektaktyczna skóra marmurowa
LIMFATYCZNE (LM)
Ograniczone / zlokalizowane
Rozlane
ŻYLNE (VM), (CVM), (AVM)
Ograniczone / zlokalizowane
Rozlane
TĘTNICZE (AM), (AVF), (AVM)
POSTACIE MIESZANE
Regionalne
Zespół Klippel-Trenaunay (CLVM)
Zespół Parkes-Weber (CLVM z AFV)
Rozlane
Zespół Maffucci (LVM, enchondroma)
Zespół Solomona (CM, VM, wewnątrzmózgowa AVM, znamiona skórne)
Zespół Riley-Smith (LVM, macrocephalia, pseudopapilledema)
Zespół Proteus (CM, VM, macrodactylia, połowiczy przerost ciała, tłuszczaki, znamiona barwnikowe, skolioza)
Malformacje kapilarne
Często są one nieprawidłowo nazywane „naczyniakami kapilarnymi”. Są to zaburzenia rozwojowe naczyń kapilarnych, a nie guzy. Istnieje kilka typów znamion naczyniowych składających się z nieprawidłowych naczyń kapilarnych.
Znamię winnej plamy
Ostro odgraniczona plamkowana zamiana typu winnej plamy jest najpowszechniejszą malformacją naczyniową. Różowe zabarwienie okresu noworodkowego z czasem ciemnieje i staje się głęboko czerwone. Zmianę należy różnicować z innymi pospolitymi znamionami plamkowymi okresu noworodkowego nazywanymi: „pocałunkami anioła”, „plamką łososiową”, lub „naevus flammeus neonatorum”.
Określenie zespołu Sturge-Webera odnosi się do malformacji kapilarnej (włosowatej), inaczej zmiany typu winnej plamy zlokalizowanej na twarzy w regionie unerwionym poprzez pierwszą gałązkę nerwu trójdzielnego, lub niekiedy obejmującym również część twarzy zapatrzoną poprzez drugą gałązkę tego samego nerwu wraz z anomaliami naczyniowymi splotu pajęczynówkowego i opony twardej po tej samej stronie. Wewnątrzczaszkowo występują zmiany o charakterze malformacji kapilarnej (włosowatej), żylnej lub tętniczo-żylnej naczyń mózgowych.
Dzieci z zespołem Sturge-Webera predysponowane są do padaczki oraz jaskry a także hipertrofii tkanek miękkich i kości, szczególnie centralnej części twarzoczaszki.
Każdy noworodek z plamą włosowatą zlokalizowaną w regionie zaopatrywanym poprzez pierwsza i drugą gałązkę nerwu trójdzielnego powinien poddany zostać wnikliwej diagnostyce w kierunku zespołu Sturge-Webera. Dzieci ze zmianami zlokalizowanymi w obrębie otrzymującym unerwienie z drugiej i trzeciej gałązki nerwu trójdzielnego nie są zagrożone powstaniem zespołu.
U dzieci z grupy ryzyka dwa razy do roku powinno być przeprowadzone badanie okulistyczne, a od trzeciego roku życia badanie okulistyczne przynajmniej raz rocznie. Jaskra występuje u 45% dzieci z zespołem Sturge-Webera i jest trudna w leczeniu. Skuteczne leczenie tak jaskry jak i padaczki uzależnione jest od wczesnego wykrycia anomalii. Pozwala to na zapewnienie dziecku właściwego rozwoju psychoruchowego.
Najlepsze obrazowanie malformacji uzyskuje się badaniem rezonansu magnetycznego, które szczególnie u dzieci starszych niż sześć miesięcy pozwala na wyraźne prześledzenie zmian wewnątrzczaszkowych, przewyższając pod tym względem tomografie komputerową.
W leczeniu malformacji kapilarnych (włosowatych) stosowany jest laser pulsacyjny barwnikowy, i to często już w okresie noworodkowym, pozwalając na uniknięcie zmian bliznowatych w obszarze poddanym laseroterapii i osiągnięcie w 50 – 70% częściowego lub całkowitego usunięcia malformacji. W szczególnych przypadkach, niepoddających się fotokoagulacji laserem, niezbędny jest zabieg chirurgiczny – wycięcie i przeszczep skóry, jeśli wymiary zmiany wykluczają pierwotne zamkniecie ubytku. Zabiegi operacyjne obejmują redukcje przerośniętych warg i dziąseł, a w okresie późniejszym inne operacyjne korygowanie kształtu twarzy.
Malformacje kapilarno-limfatyczne
Malformacje kapilarno-limfatyczne w przeszłości nazywane były znamieniem hipertroficznym, naczyniakiem brodawkowatym lub angiokeratomą.
Charakteryzują się ciemno-czerwoną, szorstką, brodawkowatą powierzchnią, a typowe zmiany obserwowane są już w chwili urodzenia. Obraz histologiczny przedstawia typowo ograniczone obszary, na które składają się rozdęte komórki skóry i tkanki podskórnej.
W warstwie brodawkowatej skóry zlokalizowane są malformacje określane jako angiokeratomy, najczęściej manifestujące się od okresu młodzieńczego. Mogą być zlokalizowane na stopach i dłoniach (a. Mibelli), kroczu (a. Fordyce), tułowiu (a. Fabry).
Postępowanie lecznicze w przypadkach zmian kapilarno-limfatycznych pojedynczych i ograniczonych polega na ich szerokim wycięciu i usunięciu tkanek aż do powięzi. O ile to możliwe zalecane jest pierwotne zamknięcie ubytku skóry lub wykonanie przeszczepu w przypadku rozleglejszych zmian. Mnogie angiokeratomy wymagają leczenia objawowego związanego ze stanami zapalnymi (antybiotyki) lub powierzchownym uszkodzeniem prowadzącym do krwawienia (elektrokoagulacja punktowych ognisk krwawienia). Rozważyć też można lasero- lub skleroterapię.
Telangiektazje
Grupa różnorodnych zmian zbudowanych z sieci poszerzonych naczyń.
Samoistna telangiektazja (uogólniona lub ograniczona) występuje tylko u kobiet, objawia się od okresu pokwitania – widoczne stają się powierzchowne poszerzenia naczyń szczególnie na kończynach dolnych.
Zespół Louis-Bar - Ataxia telangiektazja – choroba dziedziczna recesywna często doprowadzająca do śmierci przed dwudziestym rokiem życia. W wieku dwóch – trzech lat występują objawy ataksji móżdżkowej, następnie pomiędzy trzecim a szóstym rokiem życia stwierdza się zmiany naczyniowe o charakterze telangiektazji w obrębie gałki ocznej i skóry. Ponadto u pacjentów dotkniętych tą wadą stwierdzane są znaczne deficyty immunologiczne sprzyjające nawracającym stanom zapalnym dróg oddechowych i zatok.
Zespół Rendu-Osler-Weber – wrodzona telangiektazja krwotoczna jest chorobą autosomalną dominującą. Objawy mogą występować od dzieciństwa do ukończenia pokwitania. Twarz, język, nos, śluzówki ust, spojówki, dłoniowe powierzchnie palców, łożysko paznokcia, wątroba, płuca, śledziona, trzustka, mózg oraz błona surowicówkowa pokryte są dyskretnymi pajączkopodobymi zmianami. Zlokalizowane w skórze oraz błonie surowicówkowej często ulegają naciekowi zapalnemu z owrzodzeniem lub występuję z ich powierzchni krwawienia. Szczególnie trudne w leczeniu są przetoki tętniczo-żylne naczyń płucnych i wątrobowych oraz tętniaki dużych tętnic.
Cutis marmorata telegiectasia congenita – obserwowana w chwili urodzenia dziecka pod postacią siateczki ciemno fioletowych poszerzonych naczyń kapilarnych (włosowatych) i żylnych w skórze, głównie kończyny dolnej. Cienkościenne, żyło podobne zmiany mogą być spotykane w tkance podskórnej. W ciągu pierwszego roku życia postępuje stopniowa ewolucja i zmiany stają się dyskretniejsze nie mniej jednak atrofia i rozszerzenia żylne w obrębie zmiany pozostają widoczne przez całe życie.
Malformacje limfatyczne
W przeszłości nazywano te zmiany „cystic hygroma” lub „lymphangioma”. Mogą mieć charakter zlokalizowany lub uogólniony; zbudowane są z nieprawidłowych kanałów i torbieli limfatycznych. Powodują one powiększenie zajętego narządu. Malformacje limfatyczne i mieszane malformacje limfatyczno-żylne towarzyszą hipertrofii tkanek miękkich lub przerostowi układu szkieletowego.
Malformacje o charakterze torbieli wielkotorbielowatych są miękkie, gładkie i przeświecają poprzez niezmienioną skórę. Anomalie mikrotorbielowate przenikają poprzez skórę i mięśnie, a ich objawem są cienkie pęcherzyki w skórze lub mięśniach. Mogą one być zlokalizowane w miejscu dowolnego skupiska tkanki limfatycznej. Najczęściej występują na szyi i twarzy, w obrębie dołu pachowego, klatki piersiowej lub kończyn. Charakterystyczne dla torbieli limfatycznych jest powiększanie się na skutek krwawień do ich wnętrza, akumulacji płynu, lub zmian zapalnych.
Są to malformacje trudne w leczeniu. Rzadko obserwowano samoistne ich zmniejszanie się. Mogą one natomiast powodować znaczne przeszkody mechaniczne w obrębie przewodu pokarmowego i dróg oddechowych wymagają wtedy leczenia operacyjnego niekiedy w trybie nagłym. W zależności od sytuacji klinicznej może być konieczne założenie rurki tracheostomijnej lub sondy do karmienia.
Krwawienie z powierzchni opanowuje się w sposób typowy, w razie wystąpienia dolegliwości bólowych stosowane są leki przeciwbólowe, odczyny zapalne wymagają antybiotyków. Początkowo można stosować je doustnie, brak poprawy lub nasilony odczyn zapalny są wskazaniami do antybiotykoterapii dożylnej.
W przypadku zmian ograniczonych możliwe jest leczenie operacyjne i ich wycięcie. Ze względu na charakter rozprzestrzenienia się malformacji limfatycznych penetrujących pomiędzy zdrowe tkanki często konieczne jest leczenie etapowe. Nie poleca się przezskórnej skleroterapii malformacji limfatycznych wielkotorbielowatych.
Malformacje żylne
Podobnie jak malformacje limfatyczne, grupa ma bardzo szerokie spektrum objawów klinicznych od izolowanych pojedynczych poszerzeń żylnych lub mas gąbczastych do licznych zmian obejmujących wiele narządów.
W odróżnieniu od naczyniaków są miękkie i łatwo poddają się uciskowi. Często niesłusznie nazywane były naczyniaki jamistymi. Widoczne jako błękitnawe plamy o różnej intensywności barwy, malformacje mieszane żylno-kapilarne (włosowate) prezentują kolory od czerwonego do fioletowego.
U dzieci z malformacją żylną często obserwowane są zakrzepice żylne powodujące silne dolegliwości bólowe. W badaniu stwierdza się tkliwość i bolesność w miejscu wyczuwalnych zakrzepów w świetle naczynia. W badaniu radiologicznym obserwowane być mogą pojedyncze lub mnogie phlebolity. Zastój krwi w obrębie zmienionych naczyń doprowadzić może wtórnie do koagulopatii ze zużycia.
Polecanym obecnie leczeniem malformacji żylnych jest skleroterapia, poprzez bezpośrednie podawanie do zmiany środków obliterujących np. 95% alkoholu. Uzupełnieniem jest zabieg chirurgiczny w zakresie tkanek poddanych obliteracji. Zazwyczaj jednak nie jest możliwe całkowite zniszczenie malformacji żylnej. W obliteracji małych naczyń żylnych powierzchownych dobre efekty uzyskuje się laseroterapią.
Całkowitym leczeniem malformacji żylnej jest jej całkowite wycięcie, jak wspomniano uprzednio często niemożliwe do przeprowadzenia. Istotnym ograniczeniem jest efekt kosmetyczny lub czynnościowy jaki może być rezultatem zabiegu operacyjnego. Te same powody są wskazaniem do częściowej redukcji masy malformacji. Poprawa funkcji kończyny, zmniejszenie dolegliwości bólowych lub korekcja defektu kosmetycznego przemawiają za podjęciem leczenia operacyjnego.
Nie poleca się podwiązania naczynia tętniczego zaopatrującego zmianę ze względu na możliwą martwicę tkanek niedokrwionych.
Malformacje tętnicze
Jest to grupa malformacji naczyniowych charakteryzujących się szybkim przepływem krwi. Wyróżnia się kilka typów: malformacje tętnicze (AM), mieszane malformacje tętniczo-żylne (AVM) oraz przetoki tętniczo-żylne (AVF).
Przetoki tętniczo-żylne(AVF) zbudowane są z połączeń – przecieków z dużych gałęzi tętniczych do sąsiadującej żyły. Malformacje tętniczo-żylne (AVM) to ograniczone lub rozlane skupiska dysmorficznych tętnic i żył z niezliczonymi mikroskopijnymi przetokami naczyniowymi.
Leczenie malformacji jest trudne i nadal nie obowiązuje jeden „złoty standard” postępowania w tych przypadkach. Malformacje tętniczo-żylne będące źródłem dolegliwości bólowych, zmian zapalnych z owrzodzeniem, krwawień, zastoinowej niewydolności serca lub powodujące efekt masy w stosunku do sąsiadujących struktur, wymagają interwencji leczniczej. Stosowane w tym przypadku podwiązanie proksymalnego naczynia jest niebezpieczne i niejednokrotnie prowadzi do powiększenia zmiany poprzez wymuszone wypełnienie naczyń krążenia obocznego. W niektórych przypadkach skuteczne jest superselektywne zamknięcie – embolizacja zmienionych naczyń, jak i naczynia doprowadzającego. Embolizacja stosowana jest też w celu zmniejszenia dolegliwości bólowych, ograniczenia krwawienia lub zmniejszenia odczynu zapalnego. Stanowi również element leczenia skojarzonego – wraz z wycięciem zmiany prowadząc do usunięcia malformacji lub jej części. [6]
Malformacje mieszane
Mieszane malformacje naczyniowe wchodzą w skład zespołów chorobowych [12]:
- Zespól Sturge-Webera – opisany uprzednio
- Zespół Klippel-Trenaunay – współistnieje malformacja kapilarno (włosowato)-limfatyczno-żylna tułowia lub kończyn z przerostem układu kostnego (osiowym lub zwiększeniem obwodu kończyn i tyłowia)
- Zespół Parkes-Weber – objawami klinicznymi przypomina zespół Klippel-Trenaunay, a jest to kapilarno (włosowato)-limfatyczno-żylna malformacja, zwykle w obszarze kończyny górnej z przetokami tętniczo-żylnymi.
- Zespół Maffuci – żylne malformacje o egzofitycznym charakterze, wyrośla chrzęstno-kostne i enchodromatozy. Malformacje żylne znajdowane są w obrębie przewodu pokarmowego, kości lub opon mózgowo-rdzeniowych.
- Zespół Beana – zespół niebieskiego pęcherzykowego znamienia podskórnego - opisywany jako liczne często bolesne kopulaste malforamcje żylne współistniejące z anomaliami żylnymi innych narządów szczególnie przewodu pokarmowego.
- Zespól Solomana – zespół znamion naskórkowych – malformacje naczyniowe skóry i CUN współistniejące z zaburzeniami architektoniki układu kostno – szkieletowego i skupiskami znamion podskórnych.
- Zespół Riley-Smitha – mnogie żylne malformacje podskórne, rzekomy obrzęk tarczy nerwu wzrokowego oraz wielkogłowie.
- Zespół Proteus – malformacje naczyniowe zlokalizowane w skórze lub trzewiach, znamiona barwnikowe, niesymetryczny wzrost, wielkogłowie, częściowy gigantyzm stóp i dłoni, tłuszczkowatość oraz wyrośla kostne.
Leczenie farmakologiczne wskazane jest w przypadkach niektórych guzów z czynną angiogenezą, szybko rosnących i prowadzących do pogorszenia stanu chorego. Zastosowanie kliniczne znalazły: kortykosteroidy, interferon α-2a, α-2b, winkrystyna i antybiotyki.
Kortykosteriody stosowane są ogólnie lub miejscowo – drogą bezpośredniego podania w przypadku szybko powiększających się zmian naczyniowych, niejednokrotnie z odczynem zapalnym analogicznie do leczenia naczyniaków. Powodują one zahamowanie ekspansji śródtkankowej zmiany oraz ograniczenie odczynu zapalnego. Mogą także stanowić jeden z elementów leczenia skojarzonego wraz laseroterapią, chirurgiczną redukcją masy guza. Ze względu na możliwe efekty uboczne podawanie ich u dzieci wymaga ścisłej kontroli stanu pacjenta oraz monitorowania parametrów biochemicznych.
Interferon α-2a stosowano z sukcesem z przypadkach malformacji naczyniowej zlokalizowanej w obrębie tkanki płucnej, ze zmianami w przestrzeni zaotrzewnowej często powikłanej DIC lub trudnymi do opanowania chłonkotokami – jak w zespole Gorhama. Po zaprzestaniu podawania α-2a nasilały się objawy niestabilności układu krzepnięcia w tych przypadkach. Obie postacie interferonu α-2a, α-2b wykazały korzystne działanie w przypadkach naczyniaków opornych na kortykosteroidy. Aktywność ta może polegać na hamującym oddziaływaniu na zdolność tworzenia nowych naczyń, hamowaniu proliferacji lub aktywności przeciwwirusowej.
Analiza dostępnego polskojęzycznego piśmiennictwa dotyczącego zmian naczyniopochodnych u dzieci wskazuje na niespójność i niejednolitość nomenklatury dotyczącej tego zagadnienia a przede wszystkim – wynikający z tego – brak ujednoliconego schematu postępowania diagnostyczno – terapeutycznego. Konieczną wydała się zatem próba stworzenia modelu opieki i leczenia dzieci z wrodzonymi zmianami naczyniopochodnymi w oparciu o już funkcjonujące standardy na świecie i dopasowanie ich do potrzeb i realiów krajowych.
Piśmiennictwo
- Clymer M.A., Fortune D.S., Reinisch L., Toriumi D.M., Werkhaven J.A., Ries W.R. Interstitial Nd:YAG photocoagulation for vascular malformations and haemangiomas in childhood. Arch Otolaryngol Head Neck Surg. 1998;124:432-6;
- Enjolras O. Vascular Tumors And Vascular Malformations: Are We at the Dawn of a Better Knowledge? Pediatric Dermatology 1999; vol.16, No. 3: 328-241;
- Finn M.C., Glowacki J., Mulliken J.B. Congenital vascular lesions: Clinical application of a new classification. J. Pediatr. Surg. 1983; 18:894-905;
- Fishman S.J., Mulliken J.B. Haemangiomas and vascular malformations of infancy and childhood. Pediatr Clin North Am. 1993 Dec; 40(6): 1177-200;
- Frieden I., Enjolras O., Esterly N. Vascular birthmarks and other abnormalities of blood vessels and lymphatics. Plast Reconstr Surg 69: 412-422;
- Jain V, Sigh S, Pawa S, Chowdhury V. Congenital Vascular Anomalies: A Case Report and Biological Classification System. Ind J Radiol Imag 2002 12(4):527-529;
- Kern S. An investigation of haemangiomas, venous and lymphatic malformations. Acta Radiologica 41(2000): 453-457;
- Mulliken J.B., Glowacki J. Haemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg 1982; 69: 412-420;
- Vlachakis I., Gardikis S., Michailoudi E., Charissis G. Treatment of haemangiomas in children using a Nd:YAG laser in conjunction with ice cooling of the epidermis: techniques and results. BMC Pediatrics 2003;3:2-8;
- Waner M., Suen J.Y. A classification of congenital vascular lesions. Haemangiomas and vascular malformations of the head and neck.; John Wiley&Sons, Inc.; New York; 1999: 1-12;
- Waner M., Suen J.Y. The natural history of haemangiomas. Haemangiomas and vascular malformations of the head and neck.; John Wiley&Sons, Inc.; New York; 1999: 13-45;
- Wirth F.A., Lowitt M.H. Diagnosis and Treatment of Cutaneous Vascular Lesions. Am Family Physican Feb 15, 1998: 110-118;